光技術情報誌「ライトエッジ」No.32 (2009年5月発行)
2009.3 日本応用物理学会欧文誌
Photochemical Removal of SO2 and CO2
by 172nm Xe2 and 146nm Kr2 Excimer Lamps
in N2 or Air at Atmospheric Pressure
Masaharu TSUJI1,2*, Takashi KAWAHARA2, Masashi KAWAHARA2,
Naohiro KAMO2, and Nobuyuki HISHINUMA3
1 Institute for Materials Chemistry and Engineering, Kyushu University, Kasuga, Fukuoka 816-8580, Japan
2 Department of Applied Science for Electronics and Materials,
Graduate School of Engineering Sciences, Kyushu University, Kasuga, Fukuoka 816-8580, Japan
3 Ushio Inc., Himeji, Hyogo 671-0224, Japan
*E-mail address: tsuji@cm.kyushu-u.ac.jp
(Received August 10, 2008; accepted September 18, 2008; published online December 19, 2008)
The photochemical removal of SO2 and CO2 was investigated in N2 or air using 172nm Xe2(50 or 300mW/cm2) and 146 nm Kr2(25mW/cm2) excimer lamps and without using any expensive catalysts. After 30 min photoirradiation, 45 and 75% of SO2(1000 ppm) were removed in N2 and air (20% O2) at 172nm photoirradiation, respectively, whereas 39 and 8% of SO2 were removed in N2 and air at 146nm, respectively. By using a high-power Xe2 lamp, ~10 and ~45% of SO2 (200ppm) could be removed in N2 and air in a flow system, respectively. Although no photolysis of CO2 (1000ppm) was observed at 172nm, ~16 and ~8% of CO2 were converted to CO+O2 in N2 and air at 146nm after about 10min photoirradiation, respectively. Possible decomposition mechanisms were discussed.
KEYWORDS : vacuum ultraviolet photolysis, excimer lamp, 172nm, 146nm, SO2 removal, CO2 removal, environmental technology
1. Introduction
Sulfur dioxide (SO2) is formed when fuels containing sulfur, such as coal and oil, are burned, and when gasoline is extracted from oil. SO2 dissolves in water vapor to form acid and interacts with other gases and particles in the air to form sulfates and other products that can be harmful to pEOple and the environment. SO2 is a primary contributor to the formation of acid rain, which is associated with the acidification of soil, lakes, and streams, and the accelerated corrosion of buildings and monuments.1-4)
Carbon dioxide (CO2) in the Earthʼs atmosphere is a major contributor to the greenhouse effect.1-4) Its concentration in the atmosphere increased from approximately 280ppm in preindustrial times to 382 ppm in 2006 according to the National Oceanic and Atmospheric Administration, U.S.A., which is a 36% increase.2) The current rate of increase in CO2 concentration is about 1.9ppm/year. Almost all of the increase is due to human activities.
The removal of SO2 has been extensively studied using catalysts.5-7) Catalysts must be active at low reaction temperatures in the presence of such components as O2, H2O, CO, CO2, NO, and NO2, which are usually present in the exhaust gases. Recently, the cost of rare-metal catalysts has increased greatly because of economic growth in developing countries. Therefore, a new low-cost and catalyst-free removal method of SO2 in air must be developed to overcome this problem.
One catalyst-free removal technique of SO2 and CO2 is the use of electric discharge methods.8-13) In our previous study using microwave discharge without any catalysts, CO2 could be efficiently decomposed in N2 at atmospheric pressure.11) However, when CO2 diluted in N2/O2 mixtures was decomposed by microwave discharge, a large amount of NO was emitted owing to the discharge reactions of buffer N2 and O2 gases. In an electric discharge of SO2/air and CO2/air mixtures, the electron-impact excitation, ionization, and dissociation of SO2, CO2, N2, and O2 occur simultanEOusly and subsequent secondary reactions such as N(2D,2P)+O2→NO+O lead to a large amount of NO production. This NO emission is a general disadvantage when we use discharge methods for the removal of SOx, CO2, and NOx in air. Thus, we concluded that it was difficult to apply discharge methods to the removal of SO2 and CO2 in air. A higher selectivity in gas phase reactions was required to remove SO2 and CO2 efficiently in the presence of N2 and O2.
We have recently studied the photochemical removal of NOx using a 193nm ArF excimer laser without any expensive catalysts.14-17) One advantage of the photochemical method is that N2 is inert for photoirradiation above 150nm. Therefore, NO emission due to the decomposition of N2 is negligible. When ArF excimer laser photolysis is applied to the practical NOx removal process, there are many severe problems, such as the excimer laser apparatus is expensive, the running cost is high, and the apparatus is large and heavy, including high-power sources. In order to overcome these problems, we used a low-cost and compact 172nm Xe2 excimer lamp as a new vacuum ultraviolet (VUV) light source. It was applied to the photochemical removal of NO and NO2 in N2 or air in our previous studies.18-20)
In this study, we investigated the photochemical removal of SO2 and CO2 using a 172nm Xe2 excimer lamp. In addition, we also used a 146nm Kr2 excimer lamp as a new VUV source for the removal of these molecules. The conversions of each gas using these two lamps were compared. The possible removal mechanisms of SO2 and CO2 under VUV photoirradiation are discussed using the reported photochemical and gas phase kinetic data.21-24)
2. Experimental Methods
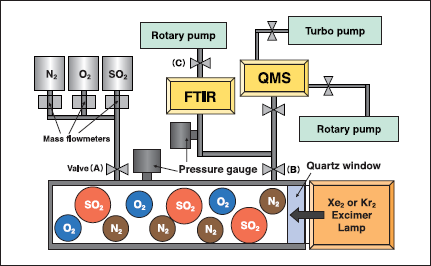
Fig.1:(Color online) Photochemical apparatus for removal of SO2 using excimer lamps. Quartz window was replaced by a MgF2 window for Kr2 excimer lamp.
Figure 1 shows the photolysis chamber with an inside volume of 185cm3 used in this study. Light from an unfocused 172 nm Xe2 excimer lamp (Ushio UER20H172 : 50 or 300mW/cm2, 155-200nm range)and a 146nm Kr2 excimer lamp (Ushio UER20H146: 25mW/cm2 120-165nm range) was used to remove SO2 and CO2 at room temperature. The low-power Xe2 and Kr2 lamps were commercial products, whereas the high-power(300mW/cm2)lamp was a trial product of Ushio. The dimensions of the low-power Xe2 (50mW/cm2) and Kr2 (25mW/cm2)lamps were Φ=30mm and length=200mm, whereas those of the high-power Xe2 lamp(300mW/cm2) were Φ=70mm and length=240mm.
Experiments were carried out not only in a closed batch system but also in a flow system. The total pressure was maintained at atmospheric pressure in most of the experiments and the SO2 and CO2 concentrations in pure N2 or air (10 or 20% O2) were 1000ppm (v/v) in the batch experiments. In the batch system, the desired gas mixtures were introduced through mass flowmeters, and valves (A) and (B) shown in Fig. 1 were closed. Then, the lamp was irradiated from the right side. In the flow system, stop valves (A)-(C)were opened during the experiments and the flow rate of each gas was controlled by a mass flow controller. The flow rate was kept at 1 L/min in the present experiments. The total pressure was maintained at atmospheric pressure using valve (B). The concentration of SO2 in the flow experiment was 200ppm.
We have also studied the O3+SO2 reaction using the same apparatus. Initially, O(3P,1D)and O3 were prepared from the 172nm photolysis of air in the photolysis chamber. After O3 was partly stored in the Fourier transform infrared (FTIR) cell, stop valve (B) was closed and the photolysis chamber was evacuated in vacuum. Then a SO2/N2 mixture was introduced to the chamber and then valve (B) was opened. Under such conditions, the short lived O(3P,1D) disappeared and only O3 with a long lifetime was present as an active species. The initial concentration of O3 in a SO2/air mixture was 1.5% and that of SO2 was 1000ppm.
In the batch experiments, before and after photoirradiation, outlet gases were analyzed by a HORIBA gas analysis system (FG122-LS) equipped with an FTIR spectrometer and an ANELVA gas analysis system (M-200GA-DTS) equipped with a quadrupole mass spectrometer. On the other hand, outlet gases were analyzed online using the FTIR spectrometer in the flow experiments. A low-sensitivity mass spectrometer was used for the determination of the N2/O2 ratios of buffergases, whereas a high-sensitivity FTIR system was used for the detection of SO2, CO, CO2, and O3. The light path length and the volume of the analyzing chamber in FTIR were 2.4m and 300 cm3, respectively. The spectra were measured in the 900-5000cm-1 region with an optical resolution of 4cm-1. The concentrations of SO2, CO, and CO2 were calibrated using standard samples supplied from gas companies. The concentration of O3 was evaluated by reference to the standard spectral data supplied by HORIBA. We determined the residual amounts of SO2 and CO2 ,[SO2]/[SO2]0 and [CO2]/[CO2]0, and the formation ratio of CO, defined as [CO]/[CO2]0, from gas analyses. Here, [SO2]0 and [CO2]0 are the initial concentrations of SO2 and CO2. The fluxes of photons in our 172 and 146nm lamps are estimated to be 4.33x1016 and 1.84x1016 photons cm-2s-1, respectively, indicating that the photon flux of the 172nm lamp is larger than that of the 146nm lamp by a factor of 2.35. When the removal rates of SO2 and CO2 and the formation rates of O3 and CO are evaluated, they are normalized to these photon fluxes of the lamps.
The following gases were used without further purification:N2(Taiyo Nissan, purity>99.9998%), O2 (Nippon Sanso, purity>99.99995%), SO2 (Taiyo Nissan, 3030ppm in high purity), and CO2 (Taiyo Nissan, 1220 ppm in high purity). SO2 and CO2 were diluted in N2 or N2/O2 mixtures before use.
3. Results and Discussion
3.1 SO2 removal in N2 or air in a batch system
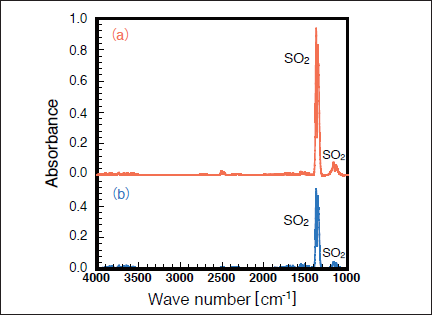
Fig.2:(Color online) FTIR spectra of SO2 (1000ppm) in N2 (a) before and (b) after 50mW/cm2 Xe2 excimer lamp irradiation for 30min.
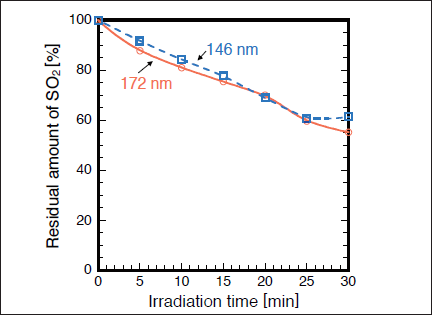
Fig.3:(Color online) Dependence of residual amount of SO2 on irradiation time in N2, when 50mW/cm2 Xe2 and 25mW/cm2 Kr2 excimer lamps were used. The initial SO2 concentration was 1000ppm.
Figures 2(a) and 2(b) show FTIR spectra of SO2 in N2 before and after 172nm photoirradiation. After photoirradiation for 30min, the SO2 peak intensities at 1360 and 1150cm-1 decreased by 45%, and no other prominent peaks were observed. Similar FTIR spectra of SO2 were obtained before and after 146nm photoirradiation. Figure 3 shows the dependence of the residual amount of SO2 on the irradiation time of 50mW/cm2 172nm Xe2 and 25mW/cm2 146nm Kr2 lamps in N2. The residual amount of SO2 at 172nm decreases almost linearly to 81, 70, and 55% with increasing irradiation time to 10, 20, and 30 min, respectively. On the other hand, the residual amount of SO2 at 146nm decreases to 84, 69, and 61% for the same irradiation times. Taking account of the photon fluxes of the two lamps, the removal rate of SO2 at 146 nm is ~2.4 times faster than that at 172 nm. Absorption cross sections of SO2, CO2, N2, O2, and O3 at 172 and 146nm are summarized in Table I. The absorption cross section of SO2 at 146nm is about 10 times larger than that at 172nm. Therefore, it is expected that the removal rate of SO2 at 146nm is about 10 times faster than that at 172nm. However, we found that the removal rate of SO2 at 146nm is only ~2.4 times faster than that at 172nm.
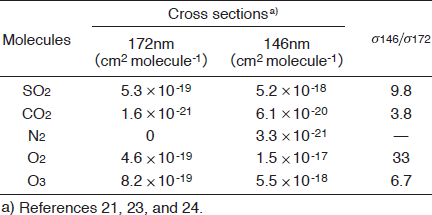
Table I : Absorption cross sections of SO2, CO2, N2, O2, and O3 at 172 and 146nm and their ratios.
At 172nm, there is no absorption of N2, whereas N2 absorbs 146nm light (see Table I). The contribution of the absorption of N2 is estimated from the following relations. The total photon energy absorbed by such a mixture of SO2/N2 while passing through the decomposition chamber, Etotal, is calculated from
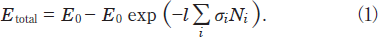
Here, E0, l, σi, and Ni are the energy of the excimer lamp, the length of the decomposition chamber, the absorption cross section of a molecule i, and its number density, respectively. The photon energy absorbed by a molecule i, Ei, is obtained from
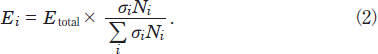
When we calculated the ESO2/(ESO2+EN2) and EN2/(ESO2+EN2) values at 146nm using known σi and Ni values, they were 61 and 39%, respectively. On the basis of this result, the decomposition rate of SO2 at 146nm is expected to be faster than that at 172nm by a factor of 6. Since this value is still larger than the observed value (~2.4), there must exist another reason for the slower removal rate of SO2 at 146nm in N2. The length of our photolysis chamber is 23cm. Incident photons loss their initial energy exponentially along the light path in the chamber owing to the absorption of N2 and attenuate to only 15% at the end of the chamber. This is another reason for the slower removal rate of SO2 at 146nm than that expected from its absorption cross section and eq. (2).
After photolysis of SO2 in N2, powders of sulfur were deposited on the walls of the reaction chamber. Thus, the dominant photolysis process for SO2 in N2 is expected to be
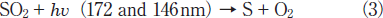
Sulfur atoms may be produced not only through direct process (3) but also through the stepwise photolysis of SO radicals.

Figure 4 shows FTIR spectra of SO2 in air (20% O2) before and after 172 nm photoirradiation. After photoirradiation for 30min, the SO2 peak intensity at 1360cm-1 decreases by 67% and we observe strong peaks of O3 at 1050, 2100, and 3060cm-1, and a weak peak due to the absorption of -SO4(e.g., H2SO4 and HSO4) groups in the 1100 ‒1200cm-1 range.25) Similar FTIR spectra of SO2 were obtained before and after 146 nm photoirradiation. Figure 5 shows the dependence of the residual amount of SO2 on the irradiation time of 50mW/cm2 172 nm Xe2 and 25mW/cm2 146nm Kr2 lamps in air. In air, a significant difference in removal rate was found between the two lamps. The residual amount of SO2 at 172nm rapidly decreases to 74, 45, and 25% with increasing irradiation time to 10, 20, and 30min, respectively. On the other hand, the residual amount of SO2 at 146nm slowly decreases to 97, 94, and 92% for the same irradiation times. Taking account of the photon fluxes of the two excimer lamps, the removal rate of SO2 at 172nm is 3.9 times faster than that at 146nm. The formation rate of O3 at 172 nm is 1.3 times faster than that at 146nm under our conditions.
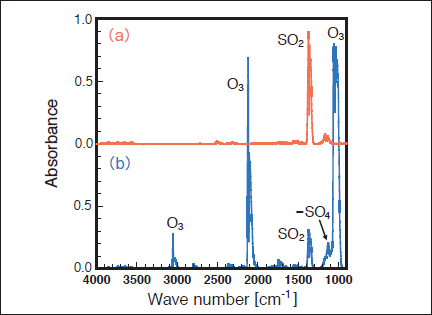
Fig.4:(Color online) FTIR spectra of SO2 (1000ppm) in air (20% O2)(a) before and (b) after 50mW/cm2 Xe2 excimer lamp irradiation for 30min.
The absorption cross sections of O2 at 172 and 146 nm correspond to 87 and 300% of those of SO2 at the same wavelengths, respectively. The concentration of O2(20% O2 in air) is 200 times higher than that of SO2. By using eq. (2), the ESO2/(ESO2+EN2+EO2), EN2/(ESO2+EN2+EO2), and EO2/(ESO2+EN2+EO2) values at 146nm were evaluated to be 0.2, 0.09, and 99.7%, respectively. On the basis of these findings, it is expected that O2 absorbs most of the photons in the initial step and gives O(3P,1D) and O3 through the following reactions.21-24)
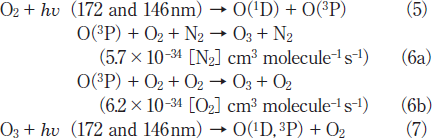
The quantum yield of O(1D) in reaction (5) is known to be 1.0 in the 140‒175nm region.24) Although the O (1D)+ SO2 reaction may contribute to SO2 removal, no information has been reported on its rate constant and products.22,23) After the photolysis of O2 and O3, the following known reactions of O(3P) and O3 with SOx and H2O are expected to occur.22,23)
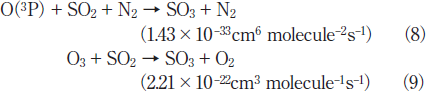
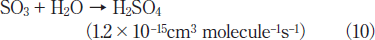
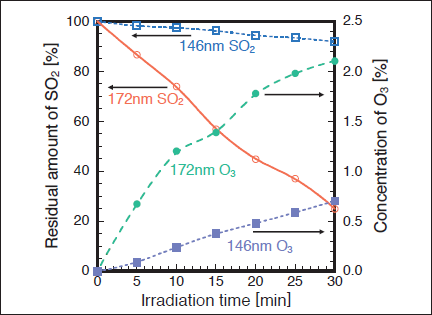
Fig.5:(Color online) Dependence of residual amount of SO2 on irradiation time in air (20% O2), when 50mW/cm2 Xe2 and 25mW/cm2 Kr2 excimer lamps were used. The initial SO2 concentration was 1000ppm.
Since the rate constant of two-body reaction (9) is very small, its relative contribution to that of three-body reaction (8) is expected to be small. When we studied the O3 + SO2 reaction using the same chamber, the residual amount of SO2 was 95.9% after 30min. This shows that reaction (9) accounts for only 4% of the total amount of SO2 removed in air. It was therefore confirmed that reaction (9) is insignificant. The H2O in reaction (10) probably originates from a small amount of residual H2O in the reaction chamber. We have recently studied NO2 removal in air using the same photolysis chamber.19) We found that all NO2 was converted to N2O5 and HNO3. It was predicted that HNO3 arose from the N2O5 + H2O reaction, where H2O was an impurity (~100ppm) in the photolysis chamber. Thus, a similar process involving the reaction of the H2O impurity, reaction (10), is expected for SO3.
The removal rate of SO2 using the 50mW/cm2 172 nm lamp in air is 3.9 times faster than that using the 25mW/cm2 146nm lamp under our conditions. It is expected that the SO2 removal in air proceeds through reactions of O(3P, 1D) with SO2 and the direct photolysis of SO2 is insignificant. Since the absorption cross section of O2 at 146nm is 33 times larger than that at 172nm, the formation rate of O3 is expected to be faster at 146 nm. However, the formation rate of O3 at 146nm is slower than that at 172nm by a factor of 1.3. The absorption cross section of O3 at 146nm is 6.7 times larger than that at 172nm. This will be one reason for the low concentration of O3 at 146nm. On the basis of these findings, one reason for the slower removal rate of SO2 at 146nm than that expected in air is the lower concentrations of O(3P, 1D) and O3 leading to O(3P, 1D) via photolysis process (7).There may be another reason for the slow removal rate of SO2 at 146nm observed in our experiment. In order to clarify it, further detailed studies and such data as rate constant and products of the O(1D)+ SO2 reaction are required.
3.2 SO2 removal in N2 and air in a flow system
For the practical application of the photochemical removal of SO2 , a flow system, in which the continuous removal of SO2 is possible, is required. We have recently found that the flow system could be applied to the removal of NO2 using the high-power 172nm Xe2 excimer lamp (300mW/cm2).19) In N2 atmosphere, ~90% of NO2 was dominantly converted to N2 and O2 under steady state conditions at an NO2/N2 mixture flow rate of 1 L/min. On the other hand, NO2 in air (5-20% O2) can be completely converted to N2O5 and HNO3.
In this study, we have also attempted to apply the flow system for the removal of SO2 using a high-power Xe2 excimer lamp because the removal rate of SO2 in the presence of O2 at 172nm is much faster than that at 146 nm. Figure 6 shows the results obtained for the SO2/N2 and SO2/air (10 and 20% O2) systems at a total flow rate of 1 L/min. The average residence time of SO2 in the photolysis chamber was estimated to be only about 0.2min. The high-power Xe2 lamp was switched on at 1min and switched off at 15min. When the excimer lamp was switched on, the residual amount of SO2 decreases to ~90 ,~80, and ~55% at O2 concentrations of 0, 10, and 20% respectively. The steady state concentrations of O3 at O2 concentrations of 0, 10, and 20% were 0, ~0.6, and ~1.1%, respectively. These results indicate that the removal rate of SO2 and the concentration of O3 increase with increasing the O2 concentration, which is consistent with the observation in the batch experiment using a low power lamp. On the basis of the present results, about one-half of the initial SO2 can be removed in air (20% O2) using the high-power Xe2 excimer lamp in our flow system even though the residence time of SO2 in the chamber is short (12s).
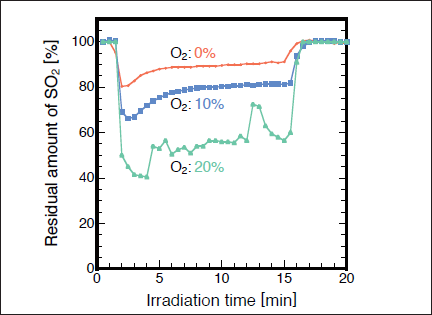
Fig.6:(Color online) Dependence of residual amount of SO2(200ppm) on irradiation time in N2 and air (10 and 20% O2) in flow system at total flow rate of 1 L/min, when 300mW/cm2 Xe2 excimer lamp was used. The lamp was switched on at 1min and switched off at 15min.
3.3 CO2 removal in N2 and air
When the high-power 172nm lamp (300mW/cm2) was irradiated to CO2 in N2 for 30min, no appreciable change in FTIR spectra was observed before and after photoirradiation. It is known that CO2 can dissociate to CO + O at 172nm.21) However, no evidence of the photolysis of CO2 was observed in this study. A major reason for this is the smaller absorption cross section of CO2 at 172nm than that of SO2 by a factor of ~330 (see Table I).
Slanger and Black26) studied the photolysis of CO2 by a 147nm lamp at low CO2 pressures below 0.25 Torr. Although they reported that CO2 can be decomposed into CO + O, it is not clear whether this technique is applicable to CO2 removal at atmospheric pressure in N2 and air. Figures 7(a) and 7(b) show FTIR spectra of CO2 before and after irradiation with the 146nm lamp in N2. The CO2 peak intensities decrease by ~20% and a very weak CO peak appears after 30min photoirradiation. Figure 8 shows the dependence of the residual amount of CO2 and the formation ratio of CO in N2 on irradiation time. The residual amount of CO2 is ~85% after only 1min photoirradiation, and then, it is nearly constant (80-85%) in the 3-30min range. This suggests that the photolysis rate of CO2 (11)becomes nearly equal to the backward reaction rate such as that of reaction (12) after 3 min photoirradiation.21-23)
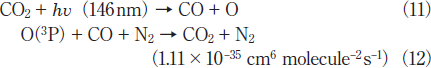
Therefore, the residual amount of CO2 becomes nearly constant above 3min.
Figures 9(a) and 9(b) show FTIR spectra of CO2 before and after irradiation with the 146nm lamp in air. The CO2 peak intensities decrease by ~8% and strong O3 peaks and a very weak CO peak appear after 30min photoirradiation. Figure 10 shows the dependence of the residual amount of CO2, the formation ratio of CO, and the concentration of O3 in air on irradiation time. The residual amount of CO2 is ~92% after 10min photoirradiation, and then, it is nearly constant (90-94%) in the 10-60min range. When the fraction of photon energy absorbed by CO2 in a CO2/N2/O2 mixture was evaluated from eq. (2), the EO2/(ECO2+EN2+EO2) value was estimated to be 99.9%. Therefore, as in the case of SO2 , the direct photolysis of CO2 is insignificant in air, and 172nm photons are almost completely absorbed by O2 in the CO2/air system. Then, O(3P, 1D) and O3 are produced through processes (5)-(7). Under such conditions, the following reactions of O(1D) with CO2, N2, and O2 occur.22,23)
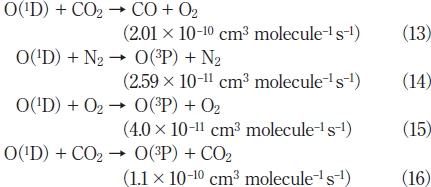
Reaction (13) is a major decomposition process of CO2 in air. The quenching reactions of O(1D) to the ground O(3P) state such as processes (14)-(16) compete with reaction (13). Since the concentration of CO2 (0.1%) is much lower than those of N2 (80%) and O2 (20%), the relative contribution of process (16) to that of processes (14) and (15) is expected to be small. After ~10min photoirradiation, the residual amount of CO2 and the formation ratio of CO become nearly constant. This shows that an equilibrium between the decomposition and formation of CO2 ↔ CO + O is established in this time range. The possible formation processes of CO2 in air are as follows.22,23)
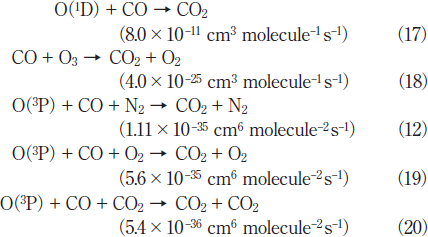
Among these reactions, two-body reaction (17) ans (12) and (19) are more significant than reactions (18) and (20), because the rate constant of reaction (18) is very small and the relative concentration of CO2 is lower than those of N2 and O2 .
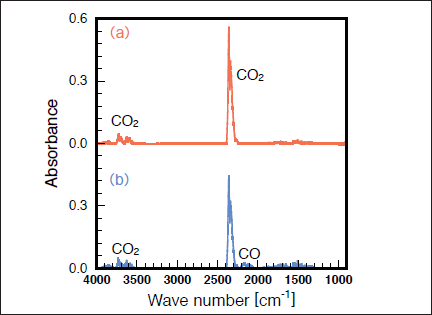
Fig.7:(Color online) FTIR spectra of CO2 (1000ppm) in N2 (a) before and (b) after 25mW/cm2 Kr2 excimer lamp irradiation for 30min.
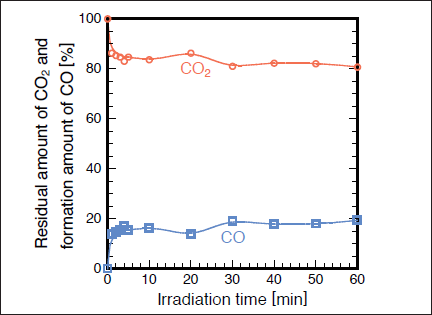
Fig.8:(Color online) Dependence of residual amount of CO2 (1000ppm) and formation ratio of CO in N2 on irradiation time, when 25mW/cm2 Kr2 excimer lamp was used.
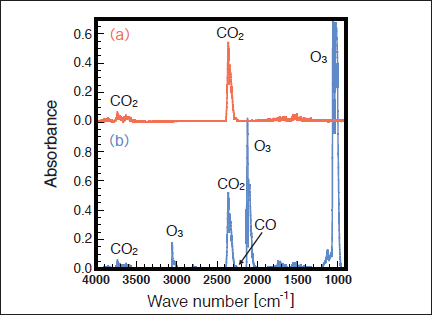
Fig.9:(Color online) FTIR spectra of CO2 (1000ppm) in air (20% O2) (a) before and (b) after 25mW/cm2 Kr2 excimer lamp irradiation for30min.
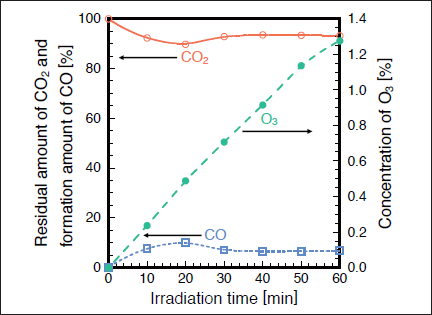
Fig.10:(Color online) Dependence of residual amount of CO2 (1000 ppm), formation ratio of CO, and concentration of O3 in air (20% O2) on irradiation time, when 25mW/cm2 Kr2 excimer lamp was used.
4. Conclusions
The photolysis of SO2 and CO2 has been studied using 172 and 146nm excimer lamps to develop a new photochemical removal process for SO2 and CO2 at an atmospheric pressure and without using any catalysts. It was found that SO2(1000ppm) can be dominantly converted to S + O2 in N2 and to ‒SO4 in air in the batch system. The conversions after 30min photoirradiation were 45% in N2 and 75% in air (20% O2) using a 50mW/cm2 172nm excimer lamp, whereas they were 39% in N2 and 8% in air (20% O2) using a 25mW/cm2 146nm excimer lamp. The major removal mechanism in N2 was the direct VUV photolysis of SO2, whereas that in air was the secondary reaction of O(3P) with SO2 followed by the SO3 + H2O reaction. These results indicated that SO2 can be removed more efficiently in the form of ‒SO4 in the presence of O2 . The removal rate of SO2 in air using the 50mW/cm2 172nm lamp was 3.9 times faster than that using the 25mW/cm2 146nm lamp because of the higher concentrations of active species such as O and O3 and the lack of absorbance of N2 at 172 nm. Therefore, we attempted SO2 removal in a flow system for practical application using a high‒power 172nm lamp (300mW/cm2). The conversions of SO2 (200ppm) were 10, 20, and 45% at O2 concentrations of 0, 10, and 20%, respectively, at a total flow rate of 1 L/min. It was found that about one-half of the initial SO2 can be removed using a flow system in air. In our apparatus with a head‒on‒type excimer lamp, light was nearly completely absorbed within only about 5mm in air due to the strong absorption of O2. To increase the removal rate of SO2 in air, we plan to carry out further experiments using a side‒on‒type excimer lamp, where light is irradiated more uniformly into the photolysis chamber.
Although no photolysis of CO2(1000ppm) was observed at 172nm photoirradiation, a small amount of CO2 was decomposed into CO + O2 at 146 nm photoirradiation. The conversions were ~16% in N2 and ~8% in air after 10min photoirradiation. For the 146nm photolysis of CO2, the backward reactions of CO such as (17), (12), and (19)suppress the high conversion of CO2 to CO + O2. In this work, we used a head‒on‒type excimer lamp. One disadvantage of this type of lamp is that the irradiation area of the lamp is restricted near the MgF2 window. Therefore, the contact area between the light and the gases is small. If a side‒on‒type 146nm excimer lamp is developed, the contact area between the light and the gases will be large; thus, a higher conversion of CO2 will be obtained. If CO is selectively removed from CO2/CO/N2/O2 gas mixtures using some absorbents, a higher conversion of CO2 by 146nm photolysis will be obtained. This is another method of improving the conversion of CO2 to CO + O2.
Acknowledgemen
This work was partly supported by NEDO and the Joint Project of Chemical Synthesis Core Research Institutions.
